
摘要:从中药新药研发的实践角度出发,围绕《基于人用经验的中药复方制剂新药临床研发指导原则》的核心内 容,结合相关的政策法规、指导原则,重点分析和讨论人用经验的内涵、基于人用经验中药新药的研发计划,并对人用 经验其他可能应用情形提出建议与思考,以期为采用人用经验进行中药新药研发的业内同仁提供参考。
为促进中医药传承创新发展,深化中药新药注 册审评体系的建设 ,加速中药新药的审批上市,相继发布了一系列文件[1-3],要求“构 建中医药理论、人用经验和临床试验相结合的中药 注册审评证据体系”(“三结合”)。国家药品监督管 理局药品审评中心(CDE)为明确“三结合”审评证据 体系的相关技术要求,于 2022 年 4 月正式发布《基 于人用经验的中药复方制剂新药临床研发指导原 则》(以下简称《指导原则》),其主要特点为充分考 虑中药的特点、研发规律和实际情况,将真实世界 研究的设计方法引入研发策略之中,针对不同的注 册分类及人用经验的收集情况,推荐相应的研发路 径[4] 。为更好地加速中药注册审评,减少研发费用及 社会资源的浪费,优选研发路径是基于人用经验中 药新药研发的关键 ,而人用经验研究的完整性、充 分性是保障研发路径选择正确、合理的基石。
笔者从中药研发的实践角度出发,以指导原则 所推荐的研发策略为核心 ,梳理相关政策法规,结 合本团队中药研发的临床实践经历,对于研发中涉 及的人用经验 、研发计划及其他可能应用的情形, 提出自己的分析与思考,供同道借鉴与参考。
1 人用经验
在“ 三结合”注册审评证据体系的概念被提出 之后,国内相关机构、业内专家相继发表了对人用 经验的认识 。孙昱认为,“ 中药是以人体实践为研发 起点,中药新药复方制剂大多具有既往临床应用经 验,即具有可供评价的人用经验”[5-6],此外还探索了 人用经验证据的分级与申报资料减免的关系 。《中 药注册管理专门规定(征求意见稿)》在“合理使用 人用经验证据”一节中,将其概括为“在长期临床实 践中积累的用于满足临床需求 ,具有一定规律性、 可重复性的关于中医临床诊疗认识的概况总结”[7]。 张晓雨等[8]认为,“ 中药人用经验不仅指在长期临床 实践中积累的关于中医临床诊疗认识的概括总结, 而且需要有一定说服力的临床数据作为支撑”,在 突出临床数据的同时,还建议应首先建立人用经验 证据的分级评价标准,以便更好地指导人用经验证 据的积累与应用。杨忠奇等[9-10]将人用经验分为人用 经验资料、人用经验数据和人用经验证据,强调基 于人用经验证据的评价 。可见,人用经验的概念和 内容相对较广泛,且认识不尽一致。
本《指导原则》将人用经验定义为“ 中药处方/制 剂在临床用药过程中积累的对其适用人群、用药剂 量、疗效特点和临床获益的认识和总结”[4],进一步 将人用经验与药品上市所需回答的关键问题相互 关联,并特别指出,其所侧重的是如何基于人用经 验产生支持监管决策的证据[4,11],即人用经验证据, 其形成过程依托于临床研究和文献研究 。各种研究类型,无论是前瞻性研究还是回顾性研究,试验性 研究还是观察性研究,均可产生支持下一步研究或 注册审评相关的证据 。人用经验证据不仅仅指有效 性和安全性,也包含与申报目标相关性和一致性的 证据。例如,可采用基于医院信息系统(HIS)的数据 挖掘,可初步获取适用人群与用法用量[12],为后续研 究的设计提供证据支持;对于同一 品种,在长期的 人用历史中,可形成丰富的人用经验证据,例如多 个适应症,多个适用人群等,建议对相关证据进行 整理,确保适用人群、干预措施、适应症与申报目标 相一致。
对于人用经验证据的评估,本《指导原则》并未 直接采用循证医学的证据分级标准 [如 GRADE 分 级[13]、牛津循证医学中心分级(OCEBM)标准等]作为 评估依据,但其仍具有重要的参考价值 。人用经验 证据的充分性,不仅要回答注册审评所需的科学问 题,以及问题的类型、多少和程度,而且还涉及拟研 发品种的社会需求问题 。不同适用病种对证据的需 求也不尽相同,对于特殊类型,例如罕见病 、危重 病,或涉及重大公共卫生的病种,或属于市场短期、 社会急需等情形,单个临床研究也可能成为强有力 的人用经验证据, 然而中药一般多用于常见疾病, 此类情况相对较少; 对于已有多种治疗选择的病 种,例如普通感冒,可能需要提供一种或多种高质 量的研究,才能形成较强的人用经验证据 。对于 3.2 类(其他来源于古代经典名方的中药复方制剂,下 同)中药而言,完善的中医理论和高质量的中医临 床实践,也可满足相应的证据需求[14]。
2 研发计划
确定临床研发计划是保障药物研发顺利实施 很为关键的内容之一 。基于人用经验中药新药的临 床研发可分为 2 个阶段,本《指导原则》根据与监管 机构沟通交流获得临床研究许可,或达成共识的时 间,分为“既往”阶段和“将来”阶段。“既往”阶段的 主要内容为人用经验临床研究,其研究类型既可以 是回顾性设计,也可有前瞻性设计,而在“将来”中, 均为前瞻性设计 。研发计划的重点在于依据“既往 ” 选择“将来”,即基于人用经验研究的信息强弱,优 选相应的研发路径 。《指导原则》以中药注册分类为 依托,推荐了 7 条临床研发路径,见表 1 。关于路径 的选择,对于申办者而言,其目的在于尽早争取注 册上市,或终止研发,从而减少资源、时间和成本的花费。
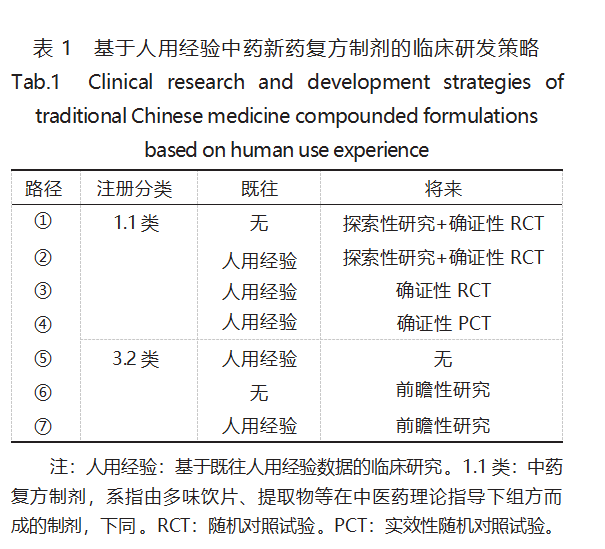
2.1 既往研究 既往研究, 也可称为人用经验研 究,即与 CDE 沟通交流前所有已完成的研究,大多 由研究者主动发起 。对于申办者而言,可以整理已 有的人用经验,直接进行沟通申请,选择相应的研 发路径,也可以继续进行人用经验研究,采用回顾 性或前瞻性的设计,补充完善人用经验证据,进而 选择相对简单的“将来”研究路径,或直接申请注 册 。对于 1.1 类 ,人用经验研究主要提供探索性证 据,对于 3.2 类而言,人用经验研究可以是探索性证 据,也可是确证性证据。
人用经验研究的设计方法可以是任何类型,对 于临床研究, 推荐采用真实世界研究的设计方法, 既符合中药临床实践应用特点,也有利于在真实医 疗环境中, 探索研发品种的临床定位及效应估计。 设计类型大致分为 A[随机对照试验(RCT)或实效 性随机对照试验(PCT)]、B(有外部对照的单臂试 验、前瞻性队列研究)、C(病例对照研究、回顾性队 列研究)、D(病例报告、病例系列、横断面研究)4 类。 对于 1.1 类中药,若选择路径③或④ , 建议人用经 验研究至少包含 B 或 A 类 。对于 3.2 类,若选择路 径⑤ , 一般来说,人用经验研究至少应为有对照的研究。
数据的可溯源性是人用经验研究的重点和关 键 。可溯源指的是可提供原始的病历记录,或源数 据库;若不可溯源,仅能提供研究的总结,可进行文 献研究,但其所能提供的证据较弱,或可作为支持 后续研究设计的证据 。此外,数据治理和质量评估 也是人用经验研究必不可少的步骤,其目的在于提 高人用经验研究的可靠性和与申报目标的一致性。
例如,在 1 项或多项人用经验研究(包括 RCT)中, 若申报目标仅为其亚组,需经过数据治理后,评估 其有效性证据,安全性可采用全部人群的结果。
2.2 将来研究 将来研究, 是与 CDE 沟通交流获 得许可后的临床研究,必须采用前瞻性设计,对于 不同的情形,须采用相应的设计方法。
对于 1.1 类,《指导原则》推荐了 4 条研发路径。 路径①,属于无人用经验的情形,本文不过多赘述。 路径② , 人用经验证据较弱,仅能回答与申报目标 相关的问题,例如适应症定位,用法用量等,可为后 续研究的设计提供证据支持,在沟通交流获得许可 后,需再进行探索性研究和确证性的 RCT 。探索性 研究的类型,可以是干预性的,也可是观察性的,根 据具体品种的情况,建议选择 RCT、PCT、前瞻性队 列研究、单臂试验中的 1 种或多种 。路径③和路径 ④相近,人用经验证据充分,结果积极或趋势明显, 能为确证性研究的设计和样本量估算提供有力支 持,可以直接开展确证性的 RCT 或 PCT 。对于选择 RCT 还是 PCT,笔者认为,若目标人群范围较广,盲 法难以开展,或适用病种复杂,目前暂无有效的治 疗措施,或中药为综合治疗方案中的 1 种,或严格控制下的 RCT 实施困难等,建议选择 PCT。此外,还 要考虑人用经验证据与后续研究类型的相关性 和一致性,例如,若已有的人用经验证据为相对严 格的临床研究,后续研究则不建议采用宽泛的 PCT研究。
对于 3.2 类,《指导原则》推荐了 3 条研发路径。 路径⑥为无人用经验证据的情形,需通过前瞻性研 究获得足够的证据支持注册 。前瞻性研究可以是干 预性的,也可以是观察性的,其将来研究的设计可 以选择前瞻性病例系列研究 、前瞻性队列研究、单 臂临床试验、RCT、PCT 中的 1 种或多种 。路径⑤和 路径⑦均为有人用经验证据的路径 ,结果可靠、证 据充分,可选择路径⑤ , 直接申请注册;证据不足, 则需进一步开展前瞻性研究,获取足够的证据支持 注册申请,设计类型可参考路径⑥。
3 其他应用情形
“ 异病同治、同病异治”是中药治疗特点之一, 随着人用经验的积累,中成药超说明书用药成为一 种常态,为更好地规范用药,上市后变更研究必不 可少 。其次,中药研发的目的也常涉及提高临床用 药的顺应性、降低用药风险、节约社会资源、保护珍 贵中药材的国家政策法规等情形,改良型新药或变更的研发需求也相对较多 。尽管本《指导原则》仅列 举了 1.1 类和 3.2 类的的研发 ,未涉及已上市中药 变更(如变更适用人群范围、变更用法用量等)和改 良型新药(如增加功能主治)等情形,考虑到《基于 “ 三结合 ”注册审评证据体系下的沟通交流指导原 则(试行)》中提到改良型新药、已上市中药变更等 情形,可参考 1.1 中药复方制剂,提出沟通交流申 请[15],笔者认为人用经验证据也可用于此类中药的 研发,建议在收集整理已有的人用经验证据后,积 极与 CDE 提出沟通交流,讨论后续研究策略 。关于 研发策略的考虑,结合相关政策法规,提出以下几 点思考。
对于 2.1 类 ,改给药途径的品种,《中药注册管 理专门规定(征求意见稿)》中要求至少应进行Ⅲ期 临床试验[7],若基于人用经验研发,建议至少进行原 给药途径对照的确证性研究,可选择路径③或④。
对于 2.3 类,增加功能主治,或上市后变更人群 范围的品种,建议参照 1.1 类和 3.2 类的研发策略, 基于人用经验证据的支持力度,优选 7 条路径中的 1 种进行实施。
对于变更用法用量的品种,根据《已上市中药 变更事项及申报资料要求》,若疗程和剂量无明显 变化,或疗程缩短、剂量降低者,至少应进行变更前 后对照的确证性临床试验;若疗程延长、剂量增加, 需按照新药处理[16]。基于人用经验研发时,前者可考 虑选择路径⑤,研究设计则采用变更前后对照的前 瞻性设计,后者建议参考 1.1 类,在路径① ~ ④中选 择 。关于儿童用法用量的细化或完善,例如“儿童酌 减”“在医师的指导下应用”等,建议优先考虑路径 ⑤~ ⑦。
对于 2.2 类改剂型 ,替代或减去有毒性或者濒 危的药味和品种,可积极开展原制剂的人用经验研 究,获得良好的数据作为对照,为后续研究的设计 提供支持[17]。
4 小结
目前,“三结合”注册审评证据体系仍处于构建 和完善的过程中 。一般认为,中医药理论主要指对 “ 理法方药”合理性的解释,是中药复方制剂临床应 用的依据,也是开展人用经验研究和(或)临床试验 的理论支撑。人用经验是中药处方/制剂在临床实践 中积累的认识与总结,本《指导原则》实际上是指可 以产生支持下一步阶段研究或监管决策的人用经 验证据 。临床试验是中、西创新药研发的必经环节,其产生的确证性证据是支持新药上市的主要依据。 中医药理论、人用经验和临床试验的互相支撑、相互 协同,可以充分利用中医药来源于临床的传统优势, 节约临床试验资源,加速中药新药的产业化进程。
本《指导原则》阐述了人用经验支持中药复方 制剂新药研发的主要原则和方法,是对中药新药研 发策略的一次变革式突破与创新 。其鼓励申办者采 用真实世界研究等方法,治理临床积累的人用经验 数据,使之形成人用经验证据,为中药新药研发提 供支持,充分彰显了中医药“ 源于临床,归于临床 ” 的特点 。本文对《指导原则》中的人用经验、研发计 划进行了分析和解读;基于中药注册的类别和品种 的特点以及人用经验证据的充分程度,提出了相应 的研发策略;对于其中未提及的注册类别,如 2.1~ 2.3 类中药改良型新药,笔者参考相关政策法规,进行了逐一分析和讨论,提出了相应的研发路径选择 策略,以供业内同仁参考。


